R语言实战:分类
目录
本文内容来自《R 语言实战》(R in Action, 2nd),有部分修改
介绍有监督机器学习领域中四种可用于分类的方法:
- 逻辑回归
- 决策树
- 随机森林
- 支持向量机
library(glue)
数据
威斯康星州乳腺癌数据集
loc <- "https://archive.ics.uci.edu/ml/machine-learning-databases/"
ds <- "breast-cancer-wisconsin/breast-cancer-wisconsin.data"
url <- glue("{loc}{ds}")
加载网络数据,并设置列名。
数据中的缺失数据用 ? 表示。
breast <- read.table(
url,
sep=",",
header=FALSE,
na.strings="?"
)
names(breast) <- c(
"ID",
"clumpThickness",
"sizeUniformity",
"shapeUniformity",
"maginalAdhesion",
"singleEpithelialCellSize",
"bareNuclei",
"blankChromatin",
"normalNucleoli",
"mitosis",
"class"
)
head(breast)
ID clumpThickness sizeUniformity shapeUniformity maginalAdhesion
1 1000025 5 1 1 1
2 1002945 5 4 4 5
3 1015425 3 1 1 1
4 1016277 6 8 8 1
5 1017023 4 1 1 3
6 1017122 8 10 10 8
singleEpithelialCellSize bareNuclei blankChromatin normalNucleoli
1 2 1 3 1
2 7 10 3 2
3 2 2 3 1
4 3 4 3 7
5 2 1 3 1
6 7 10 9 7
mitosis class
1 1 2
2 1 2
3 1 2
4 1 2
5 1 2
6 1 4
替换 class 列
df <- breast[-1]
df$class <- factor(df$class, levels=c(2, 4), labels=c("benign", "malignant"))
head(df)
clumpThickness sizeUniformity shapeUniformity maginalAdhesion
1 5 1 1 1
2 5 4 4 5
3 3 1 1 1
4 6 8 8 1
5 4 1 1 3
6 8 10 10 8
singleEpithelialCellSize bareNuclei blankChromatin normalNucleoli
1 2 1 3 1
2 7 10 3 2
3 2 2 3 1
4 3 4 3 7
5 2 1 3 1
6 7 10 9 7
mitosis class
1 1 benign
2 1 benign
3 1 benign
4 1 benign
5 1 benign
6 1 malignant
分割训练集和验证集
set.seed(1234)
train <- sample(nrow(df), 0.7*nrow(df))
df_train <- df[train,]
df_validate <- df[-train,]
table(df_train$class)
benign malignant
319 170
table(df_validate$class)
benign malignant
139 71
逻辑回归
fit_logit <- glm(
class~.,
data=df_train,
family=binomial()
)
summary(fit_logit)
lm(formula = class ~ ., family = binomial(), data = df_train)
Deviance Residuals:
Min 1Q Median 3Q Max
-2.24605 -0.08012 -0.03110 0.00266 2.11576
Coefficients:
Estimate Std. Error z value Pr(>|z|)
(Intercept) -12.4412 2.0547 -6.055 1.4e-09 ***
clumpThickness 0.7407 0.2262 3.275 0.00106 **
sizeUniformity -0.0320 0.3399 -0.094 0.92500
shapeUniformity 0.2073 0.3715 0.558 0.57680
maginalAdhesion 0.5194 0.1708 3.041 0.00236 **
singleEpithelialCellSize -0.3217 0.2613 -1.231 0.21831
bareNuclei 0.5851 0.1881 3.111 0.00187 **
blankChromatin 0.8599 0.2923 2.942 0.00326 **
normalNucleoli 0.4036 0.1828 2.208 0.02725 *
mitosis 0.8923 0.3552 2.512 0.01200 *
---
Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
(Dispersion parameter for binomial family taken to be 1)
Null deviance: 621.04 on 477 degrees of freedom
Residual deviance: 52.39 on 468 degrees of freedom
(11 observations deleted due to missingness)
AIC: 72.39
Number of Fisher Scoring iterations: 9
预测验证集,将概率转换为因子变量
prob <- predict(fit_logit, df_validate, type="response")
logit_predict <- factor(
prob > .5,
levels=c(FALSE, TRUE),
labels=c("benign", "malignant")
)
计算列联表 (混淆矩阵)
logit_perfect <- table(
df_validate$class,
logit_predict,
dnn=c("Actual", "Predicted")
)
logit_perfect
Predicted
Actual benign malignant
benign 129 6
malignant 1 69
逐步逻辑回归
生成一个包含更少解释变量的模型
fit_logit_reduced <- step(fit_logit)
class ~ clumpThickness + sizeUniformity + shapeUniformity + maginalAdhesion +
singleEpithelialCellSize + bareNuclei + blankChromatin +
normalNucleoli + mitosis
Df Deviance AIC
- sizeUniformity 1 52.399 70.399
- shapeUniformity 1 52.686 70.686
- singleEpithelialCellSize 1 53.910 71.910
<none> 52.390 72.390
- normalNucleoli 1 57.465 75.465
- mitosis 1 57.966 75.966
- blankChromatin 1 62.856 80.856
- maginalAdhesion 1 63.044 81.044
- bareNuclei 1 64.982 82.982
- clumpThickness 1 68.323 86.323
Step: AIC=70.4
class ~ clumpThickness + shapeUniformity + maginalAdhesion +
singleEpithelialCellSize + bareNuclei + blankChromatin +
normalNucleoli + mitosis
Df Deviance AIC
- shapeUniformity 1 52.876 68.876
- singleEpithelialCellSize 1 53.918 69.918
<none> 52.399 70.399
- normalNucleoli 1 57.916 73.916
- mitosis 1 58.024 74.024
- blankChromatin 1 63.272 79.272
- maginalAdhesion 1 63.735 79.735
- bareNuclei 1 64.985 80.985
- clumpThickness 1 68.728 84.728
Step: AIC=68.88
class ~ clumpThickness + maginalAdhesion + singleEpithelialCellSize +
bareNuclei + blankChromatin + normalNucleoli + mitosis
Df Deviance AIC
- singleEpithelialCellSize 1 54.154 68.154
<none> 52.876 68.876
- mitosis 1 59.402 73.402
- normalNucleoli 1 60.929 74.929
- blankChromatin 1 64.053 78.053
- maginalAdhesion 1 64.995 78.995
- bareNuclei 1 75.634 89.634
- clumpThickness 1 76.942 90.942
Step: AIC=68.15
class ~ clumpThickness + maginalAdhesion + bareNuclei + blankChromatin +
normalNucleoli + mitosis
Df Deviance AIC
<none> 54.154 68.154
- mitosis 1 60.177 72.177
- normalNucleoli 1 60.937 72.937
- blankChromatin 1 64.056 76.056
- maginalAdhesion 1 65.022 77.022
- bareNuclei 1 76.417 88.417
- clumpThickness 1 77.027 89.027
预测验证集
prob <- predict(fit_logit_reduced, df_validate, type="response")
logit_predict <- factor(
prob > .5,
levels=c(FALSE, TRUE),
labels=c("benign", "malignant")
)
计算列联表 (混淆矩阵)
logit_perfect <- table(
df_validate$class,
logit_predict,
dnn=c("Actual", "Predicted")
)
logit_perfect
Predicted
Actual benign malignant
benign 130 5
malignant 1 69
相比上述结果,精简的模型正确分类的样本比全要素模型多 1 个。
决策树
经典决策树
library(rpart)
library(rpart.plot)
使用 rpart 包中的 rpart() 函数构造决策树
set.seed(1234)
dtree <- rpart(
class~.,
data=df_train,
method="class",
parms=list(split="information")
)
print(dtree)
node), split, n, loss, yval, (yprob)
* denotes terminal node
1) root 489 170 benign (0.65235174 0.34764826)
2) sizeUniformity< 2.5 304 8 benign (0.97368421 0.02631579)
4) clumpThickness< 6.5 297 3 benign (0.98989899 0.01010101) *
5) clumpThickness>=6.5 7 2 malignant (0.28571429 0.71428571) *
3) sizeUniformity>=2.5 185 23 malignant (0.12432432 0.87567568)
6) bareNuclei< 2.5 28 13 benign (0.53571429 0.46428571)
12) sizeUniformity< 3.5 16 1 benign (0.93750000 0.06250000) *
13) sizeUniformity>=3.5 12 0 malignant (0.00000000 1.00000000) *
7) bareNuclei>=2.5 157 8 malignant (0.05095541 0.94904459) *
显示预测误差:
CP:复杂度参数nsplit:分支数rel error:训练集误差xerror:交叉验证误差xstd:交叉验证误差的标准差
dtree$cptable
CP nsplit rel error xerror xstd
1 0.81764706 0 1.00000000 1.0000000 0.06194645
2 0.04117647 1 0.18235294 0.1823529 0.03169642
3 0.01764706 3 0.10000000 0.1588235 0.02970979
4 0.01000000 4 0.08235294 0.1235294 0.02637116
绘制交叉误差与复杂度参数的关系图
plotcp(dtree)
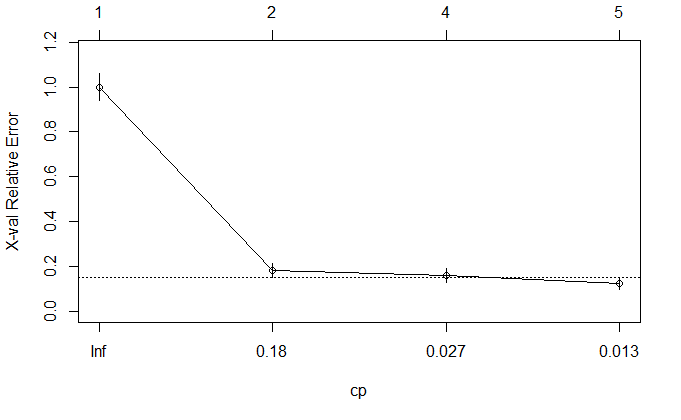
使用 prune() 函数进行剪枝
dtree_pruned <- prune(dtree, cp=0.01000000)
使用 rpart.plot 包的 prp() 函数绘制决策树
prp(
dtree_pruned,
type=2,
extra=104,
fallen.leaves=TRUE,
main="Decision Tree"
)
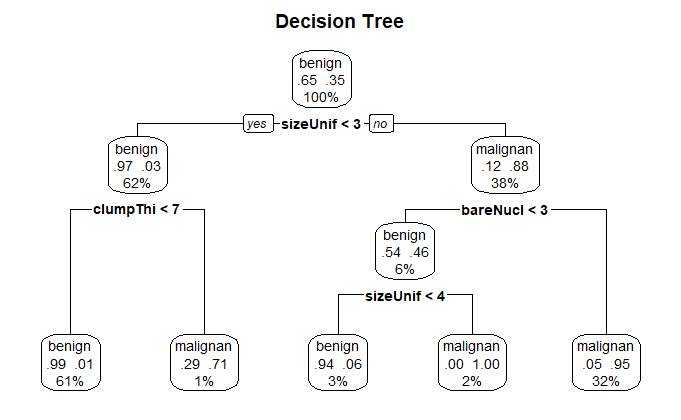
预测验证集,并计算列联表
dtree_predict <- predict(
dtree_pruned,
df_validate,
type="class"
)
dtree_pref <- table(
df_validate$class,
dtree_predict,
dnn=c("Actual", "Predicted")
)
dtree_pref
Predicted
Actual benign malignant
benign 129 10
malignant 2 69
条件推断树
条件推断树基于显著性检验选择变量和分割,而不是纯净度或同质性一类的度量
显著性检验是置换检验
library(party)
fit_ctree <- ctree(
class~.,
data=df_train
)
绘图
plot(
fit_ctree,
main="Coditional Inference Tree"
)
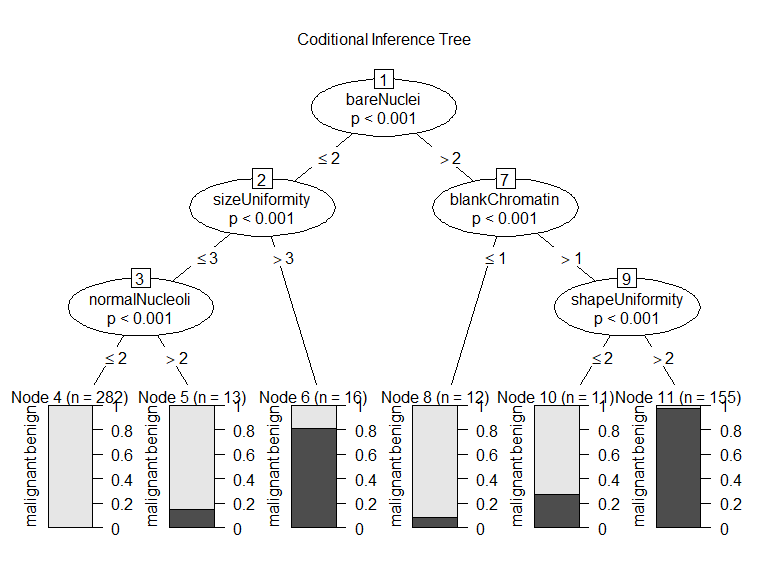
预测
ctree_predict <- predict(
fit_ctree,
df_validate,
type="response"
)
列联表
ctree_perf <- table(
df_validate$class,
ctree_predict,
dnn=c("Actual", "Predicted")
)
ctree_perf
Predicted
Actual benign malignant
benign 131 8
malignant 4 67
随机森林
library(randomForest)
使用 randomForest 包的 reandomForest() 函数生成随机森林
set.seed(1234)
fit_forest <- randomForest(
class~.,
data=df_train,
na.action=na.roughfix,
importance=TRUE
)
fit_forest
Call:
randomForest(formula = class ~ ., data = df_train, importance = TRUE, na.action = na.roughfix)
Type of random forest: classification
Number of trees: 500
No. of variables tried at each split: 3
OOB estimate of error rate: 2.66%
Confusion matrix:
benign malignant class.error
benign 313 6 0.01880878
malignant 7 163 0.04117647
变量重要性
importance(fit_forest, type=2)
MeanDecreaseGini
clumpThickness 11.382432
sizeUniformity 63.037519
shapeUniformity 49.027649
maginalAdhesion 4.275345
singleEpithelialCellSize 14.504981
bareNuclei 42.736364
blankChromatin 22.484755
normalNucleoli 11.375285
mitosis 1.755382
预测
forest_predict <- predict(
fit_forest,
df_validate
)
列联表
forest_perf <- table(
df_validate$class,
forest_predict,
dnn=c("Actual", "Predicted")
)
forest_perf
Predicted
Actual benign malignant
benign 128 7
malignant 2 68
party 包中的 cforest() 函数基于条件推断树生成随机森林
支持向量机
SVM
library(e1071)
使用 e1071 包的 svm() 函数实现
set.seed(1234)
fit_svm <- svm(
class~.,
data=df_train
)
fit_svm
Call:
svm(formula = class ~ ., data = df_train)
Parameters:
SVM-Type: C-classification
SVM-Kernel: radial
cost: 1
Number of Support Vectors: 74
预测
svm_predict <- predict(
fit_svm,
na.omit(df_validate)
)
列联表
svm_perf <- table(
na.omit(df_validate)$class,
svm_predict,
dnn=c("Actual", "Predicted")
)
svm_perf
Predicted
Actual benign malignant
benign 126 9
malignant 0 70
选择调和参数
使用带 RBF 核的 SVM 拟合样本,有两个参数:
- gamma
- cost
tune.svm() 函数使用格点搜索法
set.seed(1234)
tuned <- tune.svm(
class~.,
data=df_train,
gamma=10^(-6:1),
cost=10^(-10:10)
)
tuned
Parameter tuning of ‘svm’:
- sampling method: 10-fold cross validation
- best parameters:
gamma cost
0.01 1
- best performance: 0.02740302
使用最优参数拟合
fit_svm <- svm(
class~.,
data=df_train,
gamma=.01,
cost=1
)
预测
svm_predict <- predict(
fit_svm,
na.omit(df_validate)
)
列联表
svm_perf <- table(
na.omit(df_validate)$class,
svm_predict,
dnn=c("Actual", "Predicted")
)
svm_perf
Predicted
Actual benign malignant
benign 128 7
malignant 0 70
选择预测效果最好的解
| 统计量 | 解释 |
|---|---|
| 敏感度/正例覆盖率/召回率 | 正类的样本单元被成功预测的概率 |
| 特异性/负例覆盖率 | 负类的样本单元被成功预测的概率 |
| 正例命中率/精确度 | 被预测为正类的样本单元中,预测正确的样本单元占比 |
| 负例命中率 | 被预测为负类的样本单元中,预测正确的样本单元占比 |
| 准确率/ACC | 被正确分类的样本单元所占比重 |
评估二分类准确性
performance <- function(table, n=2) {
if (!all(dim(table) == c(2, 2)))
stop("Must be a 2 x 2 table")
tn <- table[1, 1]
fp <- table[1, 2]
fn <- table[2, 1]
tp <- table[2, 2]
sensitivity <- tp/(tp + fn)
specificity <- tn/(tn + fp)
ppp <- tp/(tp + fp)
npp <- tn/(tn + fn)
hitrate <- (tp + tn)/(tp + tn + fp + fn)
result <- paste("Sensitivity = ", round(sensitivity, n),
"\nSpecificity = ", round(specificity, n),
"\nPositive Predictive Value = ", round(ppp, n),
"\nNegative Predictive Value = ", round(npp, n),
"\nAccuracy = ", round(hitrate, n), "\n", sep="")
cat(result)
}
将 performance() 函数用于上述五个分类器
performance(logit_perfect)
Sensitivity = 0.99
Specificity = 0.96
Positive Predictive Value = 0.93
Negative Predictive Value = 0.99
Accuracy = 0.97
performance(dtree_pref)
Sensitivity = 0.97
Specificity = 0.93
Positive Predictive Value = 0.87
Negative Predictive Value = 0.98
Accuracy = 0.94
performance(ctree_perf)
Sensitivity = 0.94
Specificity = 0.94
Positive Predictive Value = 0.89
Negative Predictive Value = 0.97
Accuracy = 0.94
performance(forest_perf)
Sensitivity = 0.97
Specificity = 0.95
Positive Predictive Value = 0.91
Negative Predictive Value = 0.98
Accuracy = 0.96
performance(svm_perf)
Sensitivity = 1
Specificity = 0.95
Positive Predictive Value = 0.91
Negative Predictive Value = 1
Accuracy = 0.97
本章节均使用 0.5 作为阈值,变动阈值的影响可以通过 ROC 曲线来进一步观察
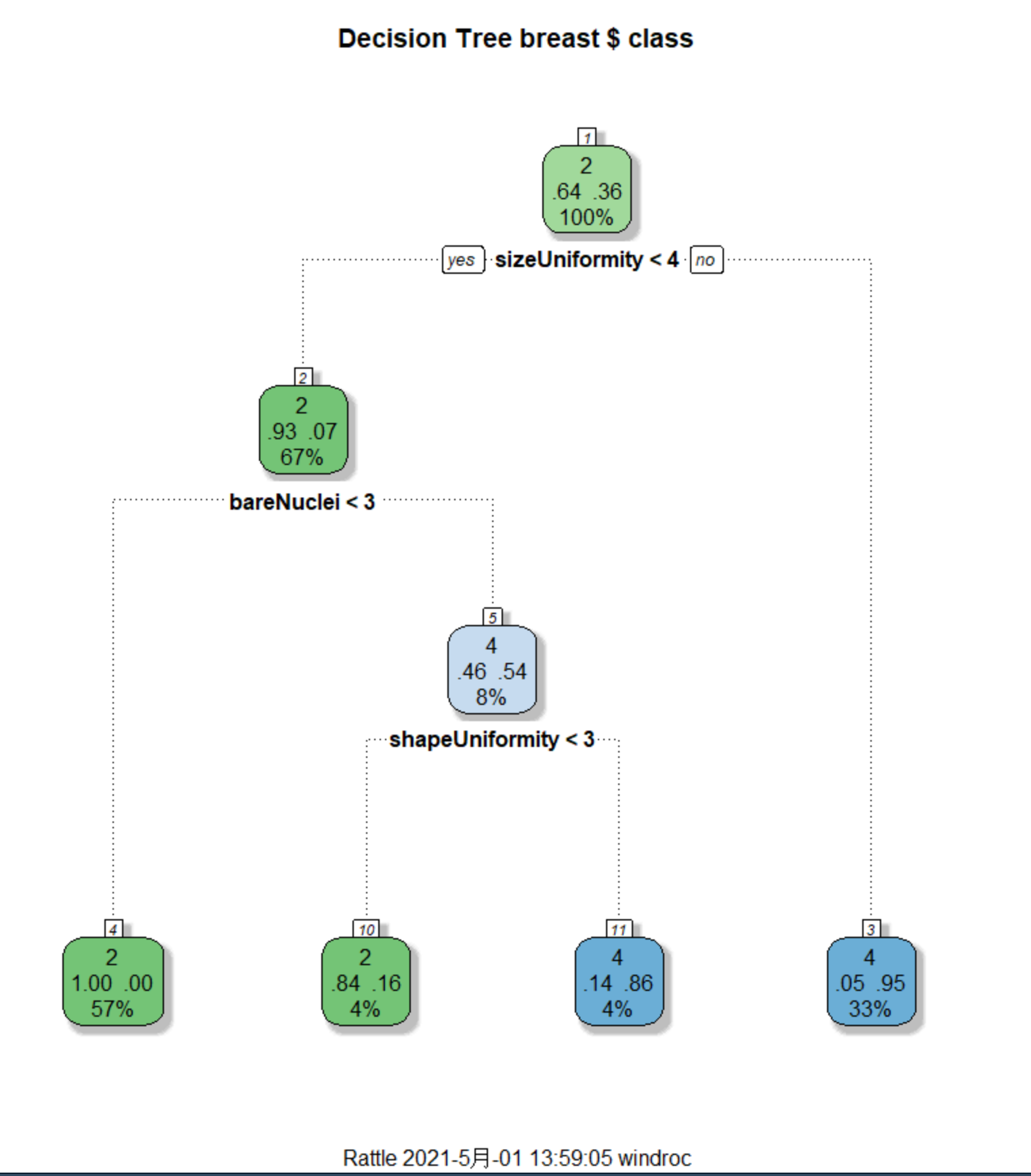
用 rattle 包进行数据挖掘
library(rattle)
rattle()
参考
https://github.com/perillaroc/r-in-action-study
R 语言实战